Словарь терминов химической кинетики и катализа
Для просмотра с помощью Microsoft Internet Explorer
Химическая кинетика (chemical kinetics) – наука
о скоростях и механизмах химических реакций.
Формальная
кинетикa – раздел
кинетики, рассматривающий временной ход реакций вне зависимости от конкретных
реагентов и продуктов.
Элементарная
реакция (elementary reaction) – протекает
без образования каких-либо веществ, отличных от продуктов либо реагентов. Как
правило, можно сказать, что преодолевается один активационный барьер.
Интермедиаты имеют время жизни менее 10-11 с.
Простая
реакция – протекает по закономерностям элементарной реакции, но
может содержать более одного активационного барьера.
Механизм
химической реакции – совокупность простых реакций (стадий), по которым
протекает реакция.
Сложные
реакции (composite reactions) – их механизм
включает две и более стадий.
Для химической реакции ![]() , стехиометрические коэффициенты ni (stoichiometric coefficient) для реагентов
(reactants) <
0, для продуктов (products) >
0.
, стехиометрические коэффициенты ni (stoichiometric coefficient) для реагентов
(reactants) <
0, для продуктов (products) >
0.
Скорость
химической реакции W (reaction rate) – число актов
превращения в единице объема V в единицу
времени.
![]() ,
,
где ni –
стехиометрический коэффициент. Если объем реагирующей системы постоянный, то
![]() .
.
Степень превращения (extent of reaction), или химическая переменная
![]() .
.
Скорость
превращения (rate of conversion) – производная по времени от степени
превращения
![]() .
.
Скорость
расходования реагента (rate of consumption) или скорость образования продукта (rate of formation):
![]() .
.
Закон
действующих масс – зависимость скорости реакции от концентраций
реагирующих частиц в виде степенной функции вида:
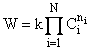 ,
,
где k –
константа скорости (rate constant, rate coefficient), ni – порядок реакции по компоненту i. Для простых реакций в газовой фазе или в
разбавленных растворах порядки реакций – целые числа, равные стехиометрическим
коэффициентам в сумме не более 3. В остальных случаях это просто удобное
выражение для математической записи скорости реакции, порядки реакции не
обязательно целые.
Порядок
реакции (overall order of reaction) – сумма порядков
реакции по всем компонентам
![]() .
.
Дифференциальное выражение для порядка
реакции по компоненту:
![]() .
.
Кинетическое
уравнение – зависимость скорости образования вещества от
концентраций реагентов. Для реакции из Nj простых
стадий
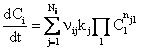 ,
,
где nij – количество частиц i, имеющее знак «+» для образующихся, «-» – для расходуемых частиц i на стадии j.
Кинетическая
кривая – зависимость концентрации или количества вещества от времени.
Основные типы элементарных реакций:
–
гомолитические – с образованием и/или разрывом электронных пар;
–
гетеролитические – разрушение и образование связей идёт без образования и
разрыва электронных пар;
– согласованные (концертные) –
синхронное превращение нескольких молекулярных орбиталей в новые МО.
Молекулярность
элементарной реакции (molecularity) – количество
частиц, участвующих в реакции. Существуют
мономолекулярные (unimolecular),
бимолекулярные (bimolecular) и
тримолекулярные (termolecular)
простые (элементарные) реакции. Простые реакции более высокого порядка не
регистрируются. Рекомбинация атомов или простых радикалов в газе –
тримолекулярная реакция с участием третьей частицы для отвода теплоты реакции.
Уравнение
Аррениуса (Arrhenius equation):
![]() ,
,
где А – предэкспоненциальный множитель (pre-exponential factor), Е –
наблюдаемая (кажущаяся) энергия активации. Хорошо описывает экспериментальные
данные при изменениях температуры менее 50 - 100 К.
Модифицированное
уравнение Аррениуса (modified Arrhenius equation) учитывает
зависимость предэкспоненциального множителя от температуры:
![]() ,
, ![]() .
.
Дифференциальное выражение для
энергии активации (activation energy):
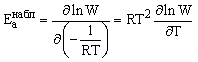 .
.
Размерности констант скорости простых
реакций: первого порядка –
с-1, второго порядка –
см3с-1, М-1с-1, третьего
порядка – см6с-1, М-2с-1.
Нормальные значения предэкспоненциального множителя:
|
мономолекулярные реакции |
1013 с-1 |
|
|
бимолекулярные реакции |
10-10 см3с-1 |
1011 М-1с-1 |
|
тримолекулярные реакции |
10-33 см6с-1 |
108 М-2с-1 |
Эффективное время реакции tэфф –
время, за которое реакция прошла бы полностью, если бы ее скорость была равна
начальной
![]() .
.
Время
полупревращения t1/2 (half life) – время, за которое концентрация реагента
достигнет арифметического среднего между начальным и конечным (равновесным)
значением.
Время жизни t (lifetime) – среднее
время существования частиц в системе:
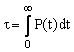 ,
,
P(t) – вероятность того, что частица не превратиться за
время t. Для
реакции первого порядка t = 1/k.
Принцип
детального равновесия (detailed balance at equilibrium, microscopic reversibility at equilibrium) – термодинамическое равновесие в
системе означает равновесие по каждой из возможных стадий каждой реакции.
Механизм обратимой реакции одинаков для прямой и обратной реакций. В
равновесии, скорость каждой стадии равна скорости обратной стадии.
Метод
стационарных концентраций (pseudo steady state assumption, steady state approximation) – это метод упрощения решения системы кинетических
уравнений, в котором принимается, что концентрация промежуточных продуктов i не меняется со временем:
![]() .
.
Прямая
задача кинетики – исходя из механизма и констант скоростей найти
зависимости концентраций от времени.
Обратная
задача кинетики – исходя из зависимостей концентраций от времени
найти механизм и константы скорости реакции.
Критерий применимости метода
стационарных концентраций:
![]() , или
, или 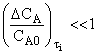 ,
,
где ![]() – стационарная концентрация промежуточного продукта,
которую находят путем приравнивания кинетических уравнений по промежуточным
веществам нулю; DCA – изменение концентрации исходного вещества А за
время установления стационарного состояния ti (transient phase) по промежуточному продукту i. Если
промежуточных продуктов несколько, то рассматривается то, для которого ti максимально.
Оценка времени достижения стационарного состояния по веществу i:
– стационарная концентрация промежуточного продукта,
которую находят путем приравнивания кинетических уравнений по промежуточным
веществам нулю; DCA – изменение концентрации исходного вещества А за
время установления стационарного состояния ti (transient phase) по промежуточному продукту i. Если
промежуточных продуктов несколько, то рассматривается то, для которого ti максимально.
Оценка времени достижения стационарного состояния по веществу i:
![]() ,
,
Wi0 –
начальная скорость образования промежуточного продукта i.
Квазиравновесное приближение - метод упрощения решения системы
кинетических уравнений, в котором принимается, что химическая реакция не
нарушает равновесие какой-либо равновесной стадии.
Условие применимости метода квазиравновесного приближения - время
установления равновесия много меньше времени реакции. tравн <<
tреакции.
Лимитирующая
стадия (rate-controlling, rate-limiting or rate-determining step) –
стадия сложной реакции, изменение константы скорости которой оказывает
максимальный эффект на скорость сложной реакции. Константа скорости
лимитирующей стадии часто входит в явном виде в выражение для скорости
образования продуктов.
Метод
псевдоизоляции состоит в добавлении большого избытка
реагента(ов), тогда его (их) концентрация постоянна; измеряется кинетика
реагента(ов) в недостатке.
Открытые
системы – обмениваются веществом с окружающей средой. Как
правило, давление можно считать постоянным.
Степень
превращения реагента в проточном реакторе x (безразмерная
величина) определяется как
![]() ,
,
где n0 – поток
вещества на входе в реактор (моль/с), n – поток вещества на выходе из реактора (моль/с).
Если в реакторе не происходит изменения скорости потока, температуры и
давления, то степень превращения
![]() ,
,
где С0 – концентрация на входе в реактор (М),
С – концентрация на выходе из реактора (М).
Время
контакта t для проточного реактора:
![]() ,
,
где Vp – объем
реактора (л), u0 –
объемная скорость подачи реакционной смеси в реактор (л/с).
Реактор
идеального (полного) смешения (РИС) (continuosly stirred tank reactor CSTR) –
идеально перемешиваемый реакционный сосуд объемом Vр (л), в
который с объемной скоростью u0 (л/с)
подается исходная реакционная смесь, содержащая реагенты с концентрацией Сi0
(моль/л). Из реактора с объемной скоростью u (л/с) выходит
конечная реакционная смесь с концентрациями Сi (моль/л).
Давление и температура одинаковы по всему объему реактора.
Реактор
идеального вытеснения – это труба объемом Vp (м3),
в которую подают с объемной скоростью u0 (м3/с)
исходную реакционную смесь, содержащую реагенты с концентрацией Сi0 (моль/м3).
Из реактора с объемной скоростью u (м3/с)
выходит конечная реакционная смесь с концентрациями Сi (моль/м3).
В трубе полностью отсутствует перемешивание, скорость движения реакционной
смеси одинакова по всему сечению трубы. Давление на входе равно давлению на
выходе.
Теория
соударений – теория расчета скорости химических реакций, в которой
предполагается, что скорость равна количеству соударений между частицами
реагентов, имеющих энергию, достаточную для преодоления активационного барьера.
Поскольку в реальности не все соударения приводят к реакции, то для расчета
скорости вводится стерический фактор
– множитель, отражающий долю соударений частиц с достаточной энергией, ведущих
к реакции.
Теория
активированного комплекса (ТАК), называемая также теорией
переходного состояния, вычисляет константу скорости реакции исходя из того, что
активированные комплексы находятся в равновесии с реагентами и превращаются в
продукты со скоростью поступательного движения вдоль координаты реакции.
Термодинамическая формулировка теории активированного комплекса
вычисляет константу скорости химической реакции исходя из термодинамических
параметров активированного комплекса – энергии Гиббса активации, энтропии
активации, энтальпии активации.
Поверхность
потенциальной энергии (ППЭ) (potential-energy surface) – потенциальная энергия молекулярного
комплекса как функция геометрических координат атомов.
Координата
реакции (reaction coordinate) – наиболее
энергетически выгодный путь от реагентов к продуктам.
Профиль
потенциальной энергии (potential energy profile) –
кривая, описывающая изменение потенциальной энергии системы атомов,
составляющих реагенты и продукты реакции, как функция координаты реакции.
Переходное
состояние (активированный комплекс, АК) (transition state, activated complex) – состояние
реагирующих частиц в точке перевала из долины реагентов в долину продуктов на
поверхности потенциальной энергии. В случае вариационной теории активированного
комплекса это состояние реагирующих частиц в точке перевала на поверхности
энергии Гиббса.
Истинная энергия активации Ea – разность энергий АК и реагентов в
невозбужденном состоянии.
Вариационная
теория активированного комплекса предполагает, что активированный
комплекс находится в точке перевала энергии Гиббса активации из долины
реагентов в долину продуктов. Положение активированного комплекса ищется путем
вычисления максимума энергии Гиббса активации или минимума константы скорости
реакции.
Туннелирование
(туннельный эффект) – пересечение барьера потенциальной энергии
частицей с кинетической энергией меньшей, чем высота барьера.
Кинетический изотопный эффект –
отношение константы скорости реакции с участием частицы, содержащей легкий
изотоп, к константе скорости реакции с участием частицы, содержащей тяжелый
изотоп:
![]() .
.
Первичный
изотопный эффект – при замене атома, смещение которого вносит
основной вклад в координату реакции.
Вторичный
изотопный эффект – при замене атома, смещение которого не вносит
существенного вклада в координату реакции.
Межмолекулярный
изотопный эффект – исходные реагенты содержат разные изотопы.
Внутримолекулярный
изотопный эффект – продукты содержат по-разному распределенные
изотопы.
Нормальный изотопный эффект наблюдается при kлегк > kтяжел.
Обратный, или
аномальный изотопный эффект наблюдается
при kлегк < kтяжел.
Клетка (cage) – агрегат из
молекул в конденсированной фазе, которые окружают фрагменты химической или
фотохимической диссоциации.
Клеточный
эффект (cage effect, Franck-Rabinowitch effect) – влияние
клеток на скорость реакций. В конденсированной среде частицы, оказавшиеся
рядом, находятся в одной клетке, вход и выход из которой затруднен:
![]() ,
,
где k-1 – константа скорости реакции в клетке
(с-1), kD –
константа скорости диффузии.
Уравнение
Бренстеда – Бьеррума:
![]() ,
,
где k0 –
константа скорости реакции А и В при всех коэффициентах активности равных 1, k – константа скорости при коэффициентах активности
реагентов и активированного комплекса gA, gB и g≠.
Первичный
солевой эффект – это изменение скорости реакции между ионами при
изменении концентрации в растворе посторонней соли, меняющей ионную силу.
Линейное соотношение свободных энергий. Изменение энергии Гиббса активации прямо пропорционально
изменению энергии Гиббса реакции:
![]() .
.
Выполняется в ряду однотипных
реакций при изменении энергии Гиббса реакции в небольшом диапазоне.
Уравнение
Брёнстеда (Brønsted) – линейная
зависимость логарифма константы скорости кислотно катализируемой реакции
от логарифма константы диссоциации
катализатора:
lg k = a lg Ka + c,
где k – константа скорости общего кислотного (основного)
катализа; Kа –
константа диссоциации кислоты (основания); a, с – эмпирические параметры для данной
реакции.
Уравнение
Гаммета (Hammett): для реакций
пара- и метазамещенных ароматических соединений
lg k = lg k0 + rs,
где k0 – константа
скорости реакции ароматического ядра без заместителей; k – константа скорости реакции ароматического ядра с
заместителем; r – эмпирическая константа, характеризующая тип
реакции; s – эмпирическая константа, характеризующая заместитель. С
точностью ±15 % позволяет
рассчитать k многих реакций.
Цепные реакции (chain reactions, initiation-propagation reactions, chain-propagating reactions) – это
химические процессы, в которых превращение исходных реагентов в продукты
осуществляется путем регулярного чередования нескольких реакций – стадий
продолжения цепи – с участием свободных радикалов. Включают обязательные
стадии:
- зарождение цепи (initiation step) – генерация
радикалов,
- продолжение цепи (propagation step, chain-propagating reaction) – проведение
реакции радикалами с сохранением свободных валентностей радикалов,
- обрыв
цепи (chain termination reaction) – исчезновение радикалов.
Также
могут содержать стадии:
- переноса цепи (transfer step) – образование
из первоначальных свободных радикалов других радикалов, которые участвуют в
стадиях продолжения цепи,
- разветвление цепи (branching) – образование
из радикалов других радикалов с большим числом свободных валентностей,
- ингибирование (inhibition) – образование
из свободного радикала малореакционноспособного продукта.
Длина
цепи n (chain length) –
число повторов стадий продолжения цепи для одного образовавшегося при
зарождении цепи радикала.
Разветвленные цепные реакции (chain branching reactions) - имеют стадию
разветвления цепи, т.е. генерации новых свободных валентностей исходя из
имеющихся. Пример – синтез воды из водорода и кислорода
Метод полустационарных концентраций Семенова. Метод
квазистационарных концентраций применяется для нахождения концентраций всех
промежуточных частиц, кроме ключевой частицы, имеющей наибольшую концентрацию
Нижний предел самовоспламенения – это концентрация (или
давление) кислорода СО2 или общая концентрация (давление) СМ,
выше которой начинается самовоспламенение. Обрыв цепи идет преимущественно на
стенке, реакция проводится вдали от мыса самовоспламенения.
Верхний предел самовоспламенения – это общая концентрация всех
частиц (давление) СМ, ниже которой происходит самовоспламенение.
Обрыв цепи идет преимущественно в объеме, если реакция проводится вдали от мыса
самовоспламенения. Существование верхнего предела для окисления водорода
обусловлено гибелью Н. в объеме смеси
Полуостров самовоспламенения – область самовоспламенения в
координатах CO2 – T или Р – Т.
Имеет мыс самовоспламенения – крайне левую точку, в которой еще
возможно самовоспламенение.
Тепловой
взрыв – неограниченный рост скорости экзотермической реакции
за счет разогрева, возникающего, если скорость тепловыделения больше скорости
теплоотдачи.
Фотохимические
реакции – это реакции, протекающие в результате поглощения
квантов ультрафиолетового, видимого или инфракрасного света. Кванты света можно
рассматривать, как индуктор.
Квантовый
выход φ (quantum yield) –
отношение числа событий к числу поглощенных системой квантов света:
![]() .
.
В случае фотохимической реакции это отношение количества прореагировавшего
реагента или образовавшегося продукта к количеству поглощенных квантов света:
![]() ,
,
где W –
скорость реакции (моль/л с); Ф – поток поглощенных квантов света (Эйнштейн/л с,
Эйнштейн – это моль квантов света).
Флуоресценция (fluorescence) –
излучательный переход из возбужденного в основное состояние без изменения
мультиплетности.
Интеркомбинационная конверсия (intersystem crossing) – безызлучательный переход между
состояниями разной мультиплетности.
Фосфоресценция (phosphorescence) – запрещенный излучательный переход в
основное состояние (например, из триплета в синглет).
Внутренняя конверсия (internal conversion) –
безызлучательный переход без изменения мультиплетности.
Тушение (quenching) возбужденного
состояния – безызлучательная
дезактивация возбужденного состояния путем взаимодействия с тушителем.
Первичные фотохимические реакции (primary photochemical reactions) – идут с участием
электронно-возбужденных молекул, приводят к изменению химической структуры и
превращению в первичные фотопродукты.
Вторичные реакции (фотохимические и темновые) – идут с участием продуктов
первичных фотохимических реакций.
Катализ (сatalysis) – явление,
состоящее в возбуждении или ускорении химических реакций под влиянием
специальных веществ – катализаторов, многократно вступающих в промежуточное
химическое взаимодействие с участниками реакции и восстанавливающих свой
химический состав после каждого цикла промежуточных взаимодействий.
Гомогенный катализ (homogeneous catalysis) – катализ, при
котором катализатор находится одной фазе с реакционной смесью.
Ферментативный катализ (enzymatic catalysis) – катализ
биологическими катализаторами – ферментами (энзимами).
Гетерогенный катализ (heterogeneous catalysis) – катализ, при
котором катализатор находится в отличной от реакционной смеси фазе.
Кислотно-основной катализ (acid-base catalysis) – катализ кислотами и основаниями.
Общий кислотно-основной катализ (general acid-base catalysis) – если скорость каталитической реакции
зависит от концентрации кислоты (основания) либо диссоциированной
(протонированной) формы.
Специфический кислотно-основной катализ (specific acid-base catalysis) – если скорость реакции зависит
преимущественно от концентраций Н+ и ОН-, либо
кислотности среды при высоких концентрациях кислоты.
Для
одного и того же механизма реакции вид кислотно-основного катализа зависит от
того, какая стадия – лимитирующая.
Функция кислотности среды Гамета:
![]() .
.
Соотношение Бренстеда для константы скорости общего кислотно-основного
катализа:
kA = a KAa,
где kA – константа
скорости реакции переноса протона, KA –
константа кислотности (диссоциации) катализатора; а и a – константы.
Конкурентное ингибирование ферментативной реакции
– замедление скорости ферментативной реакции в присутствии ингибитора In, вступающего в
конкуренцию с субстратом за связывание с активным центром фермента.
Закон действующих поверхностей:
скорость химической реакции между частицами на поверхности пропорциональна
заполнениям поверхности этими частицами (реагентами):
![]() .
.
Механизм Лэнгмюра–Хиншельвуда (Langmuir–Hinshelwood mechanism) – механизм
гетерогенной каталитической реакции, в котором образование продуктов проходит в
результате элементарных реакций между адсорбированными частицами. Данный
механизм описывает большинство гетерогенных каталитических реакций.
Механизм Или–Ридила (Eley–Rideal mechanism), ударный
механизм – это механизм гетерогенной реакции, включающий стадии взаимодействия
между адсорбированной частицей и частицей в гомогенной (газовой) фазе. Данный
механизм описывает реакции осаждения на поверхности.
Автокаталитическая реакция
катализируется одним из продуктов реакции. Например, иодирование ацетона
катализируется продуктом реакции - Н+.
![]()